
Related Publications
Molecular Structure Prediction
|
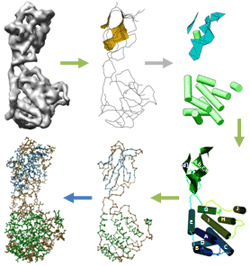
A Geometric Approach for Deciphering Protein Structure from Cryo-EM Volumes Sasakthi Abeysinghe [bib]
Electron Cryo-Microscopy or cryo-EM is an area that has received much
attention in the recent past. Compared to the traditional methods
of X-Ray Crystallography and NMR Spectroscopy, cryo-EM can be used
to image much larger complexes, in many different conformations,
and under a wide range of biochemical conditions. This is because
it does not require the complex to be crystallisable. However, cryo-EM
reconstructions are limited to intermediate resolutions, with the
state-of-the-art being 3.6A, where secondary structure elements can
be visually identified but not individual amino acid residues. This
lack of atomic level resolution creates new computational challenges
for protein structure identification. |
 |
||||
|
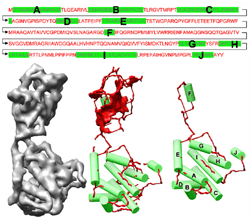
Shape modeling and matching in identifying 3D protein structures Sasakthi Abeysinghe, Tao Ju, Matthew Baker, Wah Chiu [bib]
In this paper, we describe a novel geometric approach in the process of recovering 3D protein structures from scalar volumes. The input to our method is a sequence of alpha-helices that make up a protein, and a low-resolution protein density volume where possible locations of alpha-helices have been detected. Our task is to identify the correspondence between the two sets of helices, which will shed light on how the protein folds in space. The central theme of our approach is to cast the correspondence problem as that of shape matching between the 3D volume and the 1D sequence. We model both shapes as attributed relational graphs, and formulate a constrained inexact graph matching problem. To compute the matching, we developed an optimal algorithm based on the A*-search with several choices of heuristic functions. As demonstrated in a suite of synthetic and authentic inputs, the shape-modeling approach is capable of identifying helix correspondences in noise-abundant volumes at high accuracy with minimal or no user intervention. |
 |
||||||||||
|
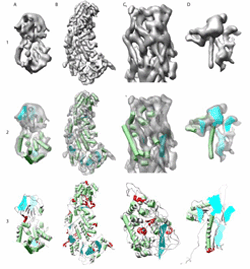
Identification of Secondary Structure Elements in Intermediate Resolution Density Maps Matthew Baker, Tao Ju, Wah Chiu [bib]
An increasing number of structural studies of large macromolecular complexes, both in X-ray crystallography and electron cryomicroscopy, have resulted in intermediate resolution (5-10 A) structures. Despite being limited in resolution, significant structural and functional information may be extractable from these maps. To aid in the analysis and annotation of these complexes, we have developed SSEhunter, a tool for the quantitative detection of alpha-helices and beta-sheets. Based on density skeletonization, local geometry calculations and a template-based search, SSEhunter has been tested and validated on a variety of simulated and authentic subnanometer resolution density maps. The result is a robust, user-friendly approach that allows users to quickly visualize, assess and annotate intermediate resolution density maps. Beyond secondary structure element identification, the skeletonization algorithm in SSEhunter provides secondary structure topology, potentially useful in leading to structural models of individual molecular components directly from the density. |
 |
||||
Geometric Skeletonization
|
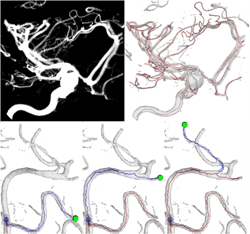
Interactive skeletonization of intensity volumes Sasakthi Abeysinghe, Tao Ju [bib]
We present an interactive approach for identifying skeletons (i.e. centerlines) in intensity volumes, such as those produced by bio-medical imaging. While skeletons are very useful for a range of image analysis tasks, it is extremely difficult to obtain skeletons with correct connectivity and shape from noisy inputs using automatic skeletonization methods. In this paper we explore how easy-to-supply user inputs, such as simple mouse clicking and scribbling, can guide the creation of satisfactory skeletons. Our contributions include formulating the task of drawing 3D centerlines given 2D user inputs as a constrained optimization problem, solving this problem on a discrete graph using a shortest-path algorithm, building a graphical interface for interactive skeletonization and testing it on a range of bio-medical data. |
 |
||||||||||
|
Segmentation-free skeletonization of grayscale volumes for shape understanding Sasakthi Abeysinghe, Matthew Baker, Wah Chiu, Tao Ju [bib]
Medical imaging has produced a large number of volumetric images capturing biological structures in 3D. Computer-based understanding of these structures can often benefit from the knowledge of shape components, particularly rod-like and plate-like parts, in such volumes. Previously, skeletons have been a common tool for identifying these shape components in a solid object. However, obtaining skeletons of a grayscale volume poses new challenges due to the lack of a clear boundary between object and background. In this paper, we present a new skeletonization algorithm on grayscale volumes typical to medical imaging (e.g., MRI, CT and EM scans), for the purpose of identifying shape components. Our algorithm does not require an explicit segmentation of the volume into object and background, and is capable of producing skeletal curves and surfaces that lie centered at rod-shaped and plate-shaped parts in the grayscale volume. Our method is demonstrated on both synthetic and medical data. |
 |
|||||||
|
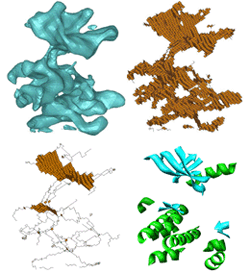
Computing a family of skeletons of volumetric models for shape description Tao Ju, Matthew Baker, Wah Chiu [bib]
Skeletons are important shape descriptors in object representation and recognition. Typically, skeletons of volumetric models are computed using iterative thinning. However, traditional thinning methods often generate skeletons with complex structures that are unsuitable for shape description, and appropriate pruning methods are lacking. In this paper, we present a new method for computing skeletons of volumetric models by alternating thinning and a novel skeleton pruning routine. Our method creates a family of skeletons parameterized by two user-specified numbers that determine respectively the size of curve and surface features on the skeleton. As demonstrated on both real-world models and protein images in bio-medical research, our method generates skeletons with simple and meaningful structures that are particularly suitable for describing cylindrical and plate-like shapes. |
 |
|||||||