
User Guide
Gorgon is a molecular modeling toolkit geared specifically toward near-atomic resolution structures from electron cryo-microscopy (cryoEM) and low-resolution X-ray crystallography. Gorgon leverages a number of computational techniques to analyze density maps, build protein backbones and refine molecular models.
The following user guide will walk you through a demo of Gorgon’s model building tools using authentic cryoEM data from one of our latest reconstructions, GroEL at 4.2Å resolution. This example will demonstrate how to build a backbone model for a portion of one GroEL subunit once you have identified the secondary structure elements using SSEHunter (available with EMAN) and have the primary sequence and secondary structure prediction for the protein. Of course, all of these files are provided in the demo. If you are interested in the overall methodology, please see our recent work on building de novo backbone traces with GroEL and Epsilon15.
Assuming you have now downloaded Gorgon `as well as the GroEL test data, let’s begin….
1. Getting started
Once you have downloaded Gorgon, unzip the files. On Windows and Mac, simply double-click on the icon for the zipped Gorgon download. For Linux, uncompress the files (tar zxvf GorgonLinux.tar.gz). Once Gorgon is uncompressed, enter the Gorgon directory. To launch Gorgon, simply click on the Gorgon icon (on Windows XP, Vista or Mac OSX Leopard) or type “./gorgon.sh” in linux.
2. The data set
The sample data set contains seven files. All but one of these files, groel-segment.seq, was created with SSEHunter and SSEBuilder, which are packaged with EMAN and have a graphical interface in UCSF’s Chimera. For more information on how to use SSEHunter and SSEBuilder, please see their user documentation. The following sections explain each of the data types and how to load them into Gorgon
Figure 1. The file open dialogue.
2.1. Volume
Volume refers to the density map; currently Gorgon only reads MRC density maps. For our GroEL example, the density map is located in the groel-demo folder and is named “densityMap.mrc”. To open the file from the menu bar, select “File>Open>Volume…”, browse to the file and open it. By default the volume is rendered in grey.
Once the volume has been opened, two new docked windows will appear. On the bottom, a volume-surface editor will allow you to alter the displayed density map. By default, the density map will be rendered as an isosurface. To adjust the displayed density threshold, slide the top slider, “Density Threshold” left to increase the isosurface displayed or to the right to decrease the displayed isosurface. The isosurface can also be controlled using the mouse wheel in combination with the control key or apple key on a Mac. The density map can be subsampled if it is too large for interactive viewing using the second slider, “Sampling Interval”. A value of ‘1’ implies no subsampling. The third slider allows the user to adjust the “Display Radius”, the amount of the density map shown relative to the selected atom or object. By default, the entire map is shown.
The second volume window, located on the right hand side of the Gorgon window, controls the display properties of the density map such as color and style. For this demo, we will utilize the “wireframe” option.
2.2 Skeleton
Skeleton refers to a skeletonized version of the density map calculated by either SSEHunter or by Gorgon. Density skeletons have two supported formats; the MRC format like the volume file or a mesh. For our GroEL example, the skeleton is located in the groel-demo folder and is named “densityMap-skeleton.mrc”. To open the file from the menu bar, select “File>Open>Skeleton…”, browse to the file and open it. By default the skeleton is rendered in red.
Like opening a volume, opening a skeleton brings up a panel in the window on the right hand side. To navigate to the window, click on the Skeleton tab located at the bottom of the right hand window. The skeleton has similar properties to the volume. For the demo, we will use the default values.
Density skeletons may also be calculated using Gorgon once a density map file has been loaded. See Section 7.2 for more information.
2.3 Helix Annotation
Helix annotation refers to the VRML representation of the helices identified by SSEHunter and SSEBuilder. For our GroEL example, the VRML helices are located in located in the groel-demo folder and named “helices-densityMap.wrl”. To open the file from the menu bar, select “File>Open>Helix Annotation…”, browse to the file and open it. Helices will be represented as green cylinders.
Opening a helix or sheet annotation will bring up a window in the right hand window that controls the display properties. For the demo, we will use the default settings.
2.4 Sheet Annotation
Sheet annotation refers to the VRML representation of the sheets identified by SSEHunter and SSEBuilder. For our GroEL example, no sheets were identified using SSEHunter and SSEBuilder. However, opening a sheet annotation file is identical to opening a helix annotation file. By default, sheet annotations will be rendered as cyan planes.
2.5 C-Alpha Atoms
C-alpha atoms are the output of Gorgon and represent the model generated using the described approach. C-alpha atoms are stored in PDB format. For the demo, no C-alpha atoms will be loaded.
2.6 Sequence and SSE Prediction
Sequence and SSE Prediction is a single text files that contains the starting residue number, the primary protein sequence and its predicted secondary structure. The file begins with “START” followed by the starting residue number. The second line contains a listing of the amino acids. The third line of this file contains the secondary structure prediction, where ‘H’ denotes a residue in a helix, ‘E’ a sheet residue and ‘-‘ which has no secondary structure assignment. For our GroEL example, the sequence file is located in the groel-demo folder and is named “groel-segment.seq”. To open the file from the menu bar, select “File>Open>Sequence and SSE Prediction…”, browse to the file and open it. This information is not immediately rendered to the screen.
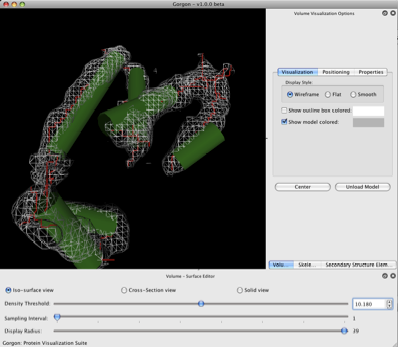
Figure 2. The groel data set.
2.7 Mouse controls
|
Left mouse |
Middle mouse |
Right mouse |
Mouse wheel |
Click |
selection |
|
|
zoom |
*Control+click |
toggle selection |
|
|
change isosurface value |
Click+drag |
rotate |
|
translate |
|
*Control+click+drag |
rotate selection |
|
translate selection |
|
Table 1. Mouse controls
*Use the Apple key instead of Ctrl in Mac systems.
3. Generating a helix correspondence
Once the data has been loaded, the next step is to calculate a correspondence between the SSEHunter determined helices and the predicted helices in the sequence. To do this, we will utilize Gorgon’s Helix correspondence finder located in the menu bar under “Actions>Secondary Structure Elements> Find Alpha-Helix Correspondences”. Selecting this option will bring up the SSE-Helix Correspondence Finder window in the right hand window. Four files are required to run the correspondence analysis, two of which are already pre-populated in the window as they were loaded in the previous steps.
3.1. Helix Lengths
The helix lengths file is another format (dejaVu) for the displayed helices. This file is a more descriptive version of the VRML helices loaded by the Helix Annotations. For the GroEL demo, the helix lengths file is located in the groel-demo folder; select the button to the right to launch a file selector and select “helix-lengths.sse”.
3.2. Sequence
The sequence file is identical to the file Sequence and SSE Prediction file that was loaded in section 2.6. If the file is not pre-populated, select the file again using the file select button to the right of this option.
3.3. Finding a correspondence
If all of the files are correctly loaded, the helices should now appear grey. Push the “OK” button to start the SSE correspondence calculation, which should take only a few seconds to run on our data set. The file selection screen of the Correspondence window should now show a list of the correspondences found by Gorgon. The various correspondences may be selected by clicking on the correspondence drop down at the top of the window. The left hand column shows the helices from the sequence while the right hand column show the matched helices from SSEHunter.
For our GroEL demo, the first correspondence appears to be the best. However for other data sets, exploring the different correspondences is advised. In fact, correspondences may be recalculated at any time by selecting “ok”. This is particularly useful if Gorgon finds some, but not all of the optimal helix pairings. These correspondences may be locked by selecting the constrained option next to the good correspondences (or by right clicking on a helix on the results table or display area) and re-running the correspondence finder with new constraints.
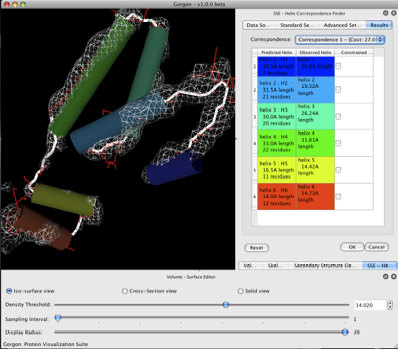
Figure 3. A helix correspondence.
4. Building a C-alpha model
After establishing the correspondence, the next step is to begin to place C-alpha atoms. To begin this process, select “Partially automated atom placement” from the menu bar under “Actions>C-alpha atoms”. This will again bring up a new window in the right hand window called “semi-automatic atom placement”. The top of this window will contain the sequence with predicted secondary structure. The helices, shown as blocks, will be colored matching the generated correspondence. Clicking on these blocks will select all the amino acids predicted to be in the helix. The amino acids below the secondary structure prediction have two states; a grey color means they are currently un-modeled (no C-alpha), while black means a C-alpha atom has been assigned in the map.
Below the sequence, the editor options can be seen. Atom editors are broken down into four classes: helix editor for placing helices, atom editor for manually placing atoms on the density skeleton, loop editor (currently disabled in the binary release) and the position editor for general atom manipulation.
For all of these editors, the default behavior can be executed by clicking on the accept button to the left of the editor tabs. The remove button will delete and un-assign any atoms assigned and selected in the sequence pane or main Gorgon viewer. Each of the editors will be explained in the following sections.
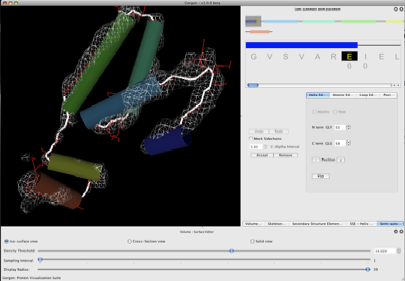
Figure 4. C-alpha atom placement.
4.1. Helix Editor
The first step in turning a correspondence into a C-alpha model is to assign atoms to the helices. Select a helix in the main viewer window by left-clicking on the cylinder. This will cause the helix to turn white as well as display the corresponding amino acids in the helix tab in the C-alpha atom placement window. In the GroEL example, the first helix (blue) should show a starting residue of 53 and an ending residue of 59.
|
The exact assignment of the helix can be shifted by using the up and down arrows located in the start and stop residue selectors. This will allow you to resolve any differences between the sequence and structure preditions. Once this is adjusted, select “Accept” and the corresponding C-alpha atoms and bonds will now be generated. Left clicking on any atom will automatically re-center the map and model about the selection; multiple selections can be accomplished using control+left-click. The residues in the sequence viewer now are black indicating that they have been assigned. Selected atoms will also be highlighted in the sequence and secondary structure views in this panel. The helix that has just been built has been arbitrarily assigned a direction, so it may be necessary to flip the direction of the helix. To flip the helix, select any atom in the helix to be flipped and then press the flip button. This will change the ordering of the helix. In the GroEL demo, the helix 1 needs to be flipped. At any time during the model building, mock sidechains may be selected. This will display relative size and biochemical properties of the amino acids. See Table 2 for a complete description. For the GroEL example, continue through the remaining helices and build C-alpha atoms for each. |
Table 2. Mock sidechain properties |
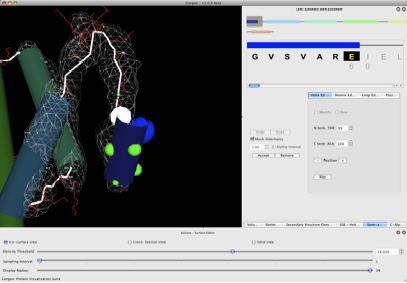
Figure 5. Helix assignment.
4.2. Position Editor
Once all of the helices have been assigned C-alpha atoms, the next step is to adjust their position and orientation in the density map. This can be accomplished in two ways. First the atoms to be rotated and translated must first be selected. This can be done in the sequence panel of the atom editor or selecting atoms in the main Gorgon window. To select multiple atoms, hold down the control key and click on the individual atoms. The first way to rotate the selected atom is to hold down the control key and the left mouse button. Dragging the mouse will cause the selection to be rotated. Translating the selection can be accomplished by simultaneously depressing the control key and right mouse button and dragging the selected atoms to their new position. For more precise control, the selected atoms can be rotated and translated from the position editor tab in the C-alpha placement window. Moving the slider or clicking on the ‘+’ or ‘-“ buttons will perform the desired operation on the selected atoms.
For the GroEL demo, helix 2 is too long and must be translated to fit within the density map. Using the mock side chains option, it is also possible to identify the large, bulky residues and match those residues up with the observed density protrusions by rotating and translating the helices. In helix 4, the density map curves slightly and as such, the two halves of the helix must be fit independently. This can be done by selecting atoms from the first half of the helix, adjusting the fit, then repeating the process for the other half of the helix.
4.3. Loop Editor
At this time, the loop editor is not enabled.
4.4. Atom Editor
|
Once the helices have been correctly positioned in the density map, it is then time to build in the loops and connect the helices. This is accomplished with the atom editor tab in the atom placement window. The atom editor will add consecutive atoms in either a forward or reverse direction from any currently assigned atom. The atom editor identifies potential positions on the density skeleton to place new atoms based on the C-alpha interval. The default C-alpha interval is set to 3.8Å, but in several parts of the GroeEL density map, this interval must be changed in order to fit the required number of atoms in loops connecting the helices. C-alpha positions can be later adjusted as described in section 4.2. |
Figure 6. Atom editor |
To use the atom editor, select a single atom that is unconnected to another consecutive atom. At the top of the atom editor tab, a direction can be selected. If you want to assign increasing consecutive atoms, select the forward direction. Conversely assigning decreasing consecutive amino acids, select the backwards direction. The arrows at the bottom of the tab will increment the selection one atom at a time in the given direction. When an atom is reached that is unassigned, possible positions for the amino acid will be shown in the main Gorgon viewer and can be selected using the spinner in the atom editor tab. Possible atom positions are shown in green while the currently selected atom position is shown in blue. To accept and place the atom, press ‘Accept’. For the GroeEL demo data, continue this of remaining atoms until the assignment is complete.
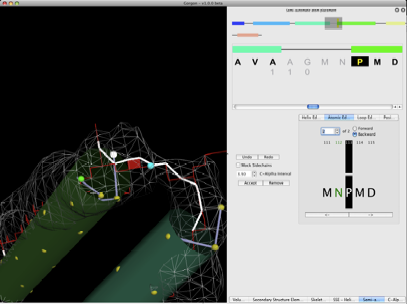
Figure 7. Atom placement. The currently selected residue is P113. Moving towards the N-terminus (backwards), the atom editor has found two possible positions for N112 (in green in the atom editor window) on the skeleton that 3.5Å from the P113. The currently selection is show in blue. Pressing accept sets the position of this atom and automatically decrements one residue when in the backward direction or increments one residue when forward is selected. The process is repeated until the loop is completed.
5. Undo/Redo
In the atom placement window, the undo and redo buttons will undo or redo atom placement and movement. Undo and redo will not work on helix placement or assignment.
6. Saving Data
Once a model has been created in Gorgon, the model can then be saved or exported as a PDB file. Saving the C-alpha model will save all assigned positions as well as create placeholders for the unassigned residues. This type of file may be loaded as a C-alpha file from the “File>Open>C-Alpha Atoms” option. An exported model file contains only the atoms assigned in Gorgon.
7. Additional Actions
Gorgon contains a number of additional utilities that were not utilized in the GroEL demo. These actions are locate under the Actions menu.
7.1. Volumes
These actions all operate on the volume data set.
7.1.1. Normalize
This allows the user to normalize a density map such that its density values are between 0 and 1
7.1.2. Downsample
Reduces the size of a density map by half in all dimensions. This is useful when working with very large volumes.
7.1.3. Skeletonization
The density map can be skeletonized by one of three methods:
Binary Skeletonization: This performs skeletonization on the density map given a user-specified density threshold. This is the method currently used by SSEHunter. More information can be found in the paper: Computing a family of skeletons of volumetric models for shape description
Grayscale Skeletonization: This method does not require a user-specified density threshold, and provides better results when all object features cannot be captured at any single density threshold. More information can be found in the paper: Segmentation-free skeletonization of grayscale volumes for shape understanding
Interactive Skeletonization: This method provides a easy to use interface which allows the user to quickly sketch the skeleton of a given density volume. Although this requires a small time investment (most often under 10 mins) by the user, it can be used to create visually accurate skeletons even in very noisy data sets. More information can be found in the paper: Interactive segmentation-free skeletonization of Grayscale Volumes
Interactive Skeletonization Instructions:
Open a volume: File -> Open -> Volume
Start interactive skeletonization: Actions -> Volume -> Skeletonization -> Interactive
Click Start
To create a new segment:
1. Hold Ctrl* and click to place a new seed
2. Hold Ctrl* and move mouse to create path from seed or
Hold Alt and move mouse to create branch from seed or
Hold Shift and move mouse to paint preferred path from seed
3. Continue holding Ctrl* or Alt and click to complete segment
4. Repeat steps 2 and 3
5. Press Esc to finish skeletonization
To edit an existing segment:
1. Click to select beginning of segment
2. Click to select end of segment
3. Press Del to delete the segment
Button |
Press |
Hold |
Hold and Click |
Ctrl* |
|
enter polyline mode |
place new seed |
Alt |
|
enter branch mode |
place new branch |
Shift |
|
enter sketch mode |
|
Left Mouse |
begin or end selection |
|
|
Del |
delete current selection |
|
|
Esc |
finish current segment |
|
|
Table 2. Interactive Skeletonization Controls
*Use the Apple key instead of Ctrl in Mac systems.
7.1.4. Crop
Crop brings up a window that will allow the user to crop the volume in the X, Y and Z directions.
7.2. Skeletons
These actions all operate on the density skeleton.
7.2.1. Laplacian smoothing
This option will smooth the skeleton, removing the “jaggy” edges in the skeleton.
7.3. Secondary structure elements
These actions all operate on the secondary structure elements.
7.3.1. Find Alpha-helix correspondence
This option is described in detail in Section 3.3.
7.4. C-alpha atoms
These actions all operate on the secondary structure elements.
7.4.1. Partially automated atom placement
This option is described in detail in Section 4.
7.4.2. Manual C-alpha atom placement
This is a free form editor that allows the placement of C-alpha atoms anywhere on the density skeleton. Left-clicking on the density skeleton will access the coordinates on the skeleton and populate the X,Y and Z positions in the Manual atom placement window. Additional information can be entered at this time. The atom can then be placed on the skeleton by clicking on the “Add Atom” button.
8. Window
The window menu displays all the windows in Gorgon. Currently open windows are denoted by a check mark. Selecting or deselecting the window title in this menu will show or hide the window.
9. Themes
Themes are preset lighting and viewing conditions. Full scene control can be set using the “Scene options” window located in the “Window” menu. New themes can be saved and loaded in subsequent sessions of Gorgon.
10. Help
Help is not currently active. However, the help menu has links that allows the user to report bugs, get updates, find related publications and get license information.